Introduction: More Than Just a Molecule of Relaxation
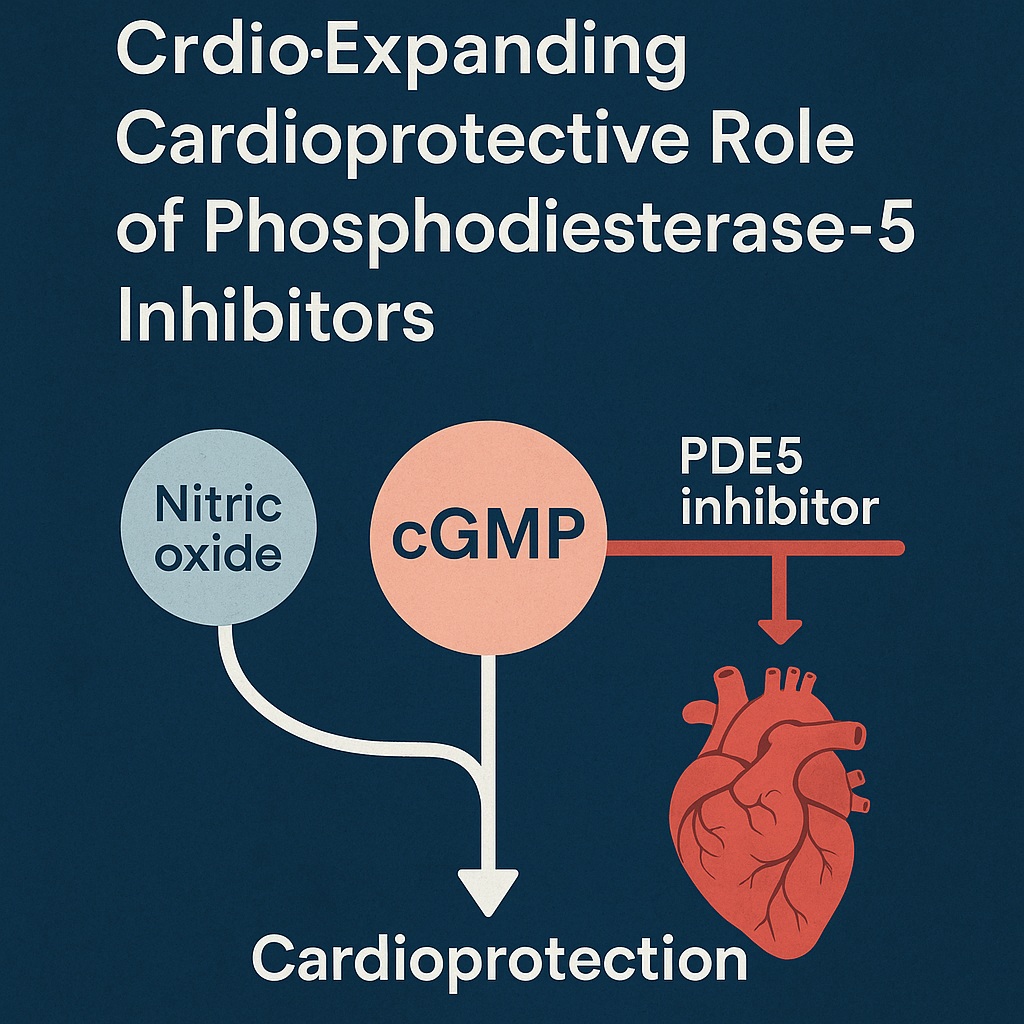
Cyclic guanosine monophosphate (cGMP) has long been known as a second messenger critical to vascular smooth muscle relaxation, a discovery that ultimately revolutionized the management of erectile dysfunction through the development of sildenafil. Yet the reach of this humble nucleotide extends far beyond facilitating vasodilation. It plays a decisive role in regulating cardiac growth, remodeling, contractility, and resistance to injury. When manipulated pharmacologically, cGMP signaling represents a therapeutic lever for protecting the heart against ischemia, hypertrophy, and even the toxic effects of chemotherapy.
Phosphodiesterase-5 (PDE5) inhibitors such as sildenafil, tadalafil, and vardenafil were initially designed to prolong cGMP signaling in penile smooth muscle. But over the last two decades, accumulating evidence has demonstrated that their influence reaches the myocardium itself. From reducing infarct size during ischemia-reperfusion injury to reversing maladaptive remodeling in heart failure, PDE5 inhibitors are redefining their place in cardiology. As clinical research advances, the notion that sildenafil might someday be as valuable in the coronary care unit as in the bedroom becomes less fanciful.
In this article, we will explore how cGMP signaling modulates cardiac health, review the cardioprotective mechanisms of PDE5 inhibition, and discuss promising avenues in conditions ranging from Duchenne muscular dystrophy to doxorubicin cardiotoxicity. Along the way, we will also consider the ironies of drug repurposing: how a treatment originally dismissed as a “lifestyle enhancer” is steadily climbing the ranks of serious cardiovascular therapeutics.
The Molecular Backbone: Regulation of cGMP by Phosphodiesterases
The biological effects of cGMP are tightly regulated by the action of phosphodiesterases, enzymes responsible for degrading cyclic nucleotides. Among the 11 PDE families identified, PDE5 is particularly specific for cGMP. By hydrolyzing this molecule into GMP, PDE5 effectively dampens its signaling potential. Thus, inhibition of PDE5 represents an elegant strategy to amplify cGMP bioavailability.
While PDE5 is classically abundant in vascular smooth muscle, its expression in the myocardium has been a subject of debate. Early reports suggested an absence in normal cardiomyocytes, but subsequent studies identified PDE5 upregulation in hypertrophied right ventricles and failing left ventricles. The implication is profound: increased PDE5 activity may contribute to maladaptive remodeling and impaired contractility. Consequently, PDE5 inhibitors like sildenafil can restore balance by preventing excessive cGMP breakdown, preserving signaling that is otherwise lost in diseased hearts.
It is worth noting that cGMP does not act in isolation. Its interplay with cyclic adenosine monophosphate (cAMP) introduces layers of complexity. PDE1 and PDE3, for example, have dual substrate specificity, allowing for cross-talk between cAMP and cGMP pathways. This biochemical dance underscores the need for precision when designing therapies that manipulate cyclic nucleotide metabolism. Nonetheless, the selective potency of PDE5 inhibitors offers a clinically tractable means of enhancing cGMP without broad, unpredictable consequences.
cGMP in Ischemic Pre- and Post-Conditioning
The phenomenon of ischemic pre-conditioning—where brief, non-lethal ischemia renders the heart more resistant to a subsequent insult—relies heavily on cGMP signaling. Nitric oxide (NO), through activation of soluble guanylyl cyclase (sGC), generates cGMP, which in turn activates protein kinase G (PKG). PKG then modulates mitochondrial and sarcolemmal channels to stabilize ionic homeostasis during stress, limiting calcium overload and preventing catastrophic cell death.
Animal models have convincingly demonstrated that enhancing cGMP at the time of reperfusion mimics the protective effect of ischemic conditioning. For instance, bradykinin and natriuretic peptides exploit this pathway to reduce infarct size. Similarly, pharmacological agents like cinaciguat—a direct activator of sGC—achieve comparable results by elevating cGMP independently of NO availability. These findings highlight that cGMP is not merely a downstream mediator but a central determinant of myocardial survival during ischemia-reperfusion.
Here enters sildenafil. When administered prior to ischemia, sildenafil reduced infarct size in experimental models by opening mitochondrial ATP-sensitive potassium channels and activating PKG. Notably, this effect was independent of its systemic hypotensive action, suggesting a direct myocardial benefit. Moreover, even when infused at reperfusion, sildenafil conferred substantial protection, positioning it as a pharmacological surrogate for ischemic post-conditioning. What was once a serendipitous side effect has thus emerged as a deliberate therapeutic tool.
cGMP-Modulatory Drugs and the Expanding Arsenal
While PDE5 inhibitors have captured much of the spotlight, they are part of a broader family of cGMP-modulatory agents. Natriuretic peptides such as atrial (ANP) and B-type (BNP) peptides activate particulate guanylyl cyclase, leading to cGMP accumulation with antihypertrophic and antifibrotic consequences. Clinical use of recombinant BNP (nesiritide) for heart failure, however, has been tempered by mixed efficacy and safety outcomes.
Cinaciguat, by contrast, has garnered attention for its ability to activate oxidized or heme-free sGC—a condition often encountered in ischemic tissues. Preclinical studies suggest that cinaciguat protects against reperfusion injury by engaging PKG-dependent hydrogen sulfide signaling. Although not yet widely adopted, this drug represents an intriguing adjunct or alternative to PDE5 inhibition in restoring cGMP signaling under oxidative stress.
Nevertheless, it is PDE5 inhibitors—sildenafil, tadalafil, and vardenafil—that remain the most clinically accessible agents. Their pharmacokinetics vary, with tadalafil’s longer half-life offering up to 36 hours of activity. Yet their unifying feature remains the same: selective amplification of cGMP signaling. This consistency allows them to be studied across diverse cardiac models, from ischemic injury to heart failure, with reproducible protective outcomes.
Sildenafil and the Fight Against Cardiac Hypertrophy
Chronic pressure overload, as seen in hypertension and valvular disease, drives the myocardium into hypertrophic growth. While initially adaptive, this process ultimately predisposes to arrhythmias, fibrosis, and failure. cGMP signaling serves as a natural brake on hypertrophic remodeling, countering pathways like calcineurin-NFAT that promote pathological growth.
Sildenafil exerts its antihypertrophic effects through PKG activation, which influences regulators of G-protein signaling and modulates calcium-handling channels. In murine models, sildenafil administration prevented the progression of pressure-overload hypertrophy without reducing afterload. This observation confirms that its action is myocardial-specific rather than a byproduct of systemic vasodilation. Importantly, chronic PDE5 inhibition also mitigated left ventricular remodeling following myocardial infarction, a context where maladaptive hypertrophy accelerates heart failure.
The clinical implications are promising. Heart failure with preserved ejection fraction (HFpEF), characterized by stiff hypertrophied ventricles, remains notoriously difficult to treat. Trials such as RELAX (Evaluating the Effectiveness of Sildenafil in HFpEF) were designed to test whether augmenting cGMP signaling translates into improved exercise capacity and outcomes. Although results have been modest, the mechanistic rationale persists: by reversing maladaptive remodeling, sildenafil may find its niche in carefully selected subgroups of patients.
Duchenne Muscular Dystrophy: A Genetic Disorder with a Cardiac Angle
Duchenne muscular dystrophy (DMD) is a devastating X-linked disorder marked by progressive skeletal and cardiac muscle degeneration. Loss of dystrophin disrupts nitric oxide synthase localization, diminishing NO-cGMP signaling and predisposing to cardiomyopathy. As skeletal muscle deteriorates, cardiac dysfunction emerges as a major cause of morbidity and mortality.
Here again, sildenafil demonstrates utility. In dystrophin-deficient mice, chronic sildenafil administration restored diastolic function and reversed established cardiomyopathy within days. This rapid improvement underscores the centrality of cGMP signaling in maintaining cardiac integrity in DMD. Importantly, sildenafil achieved these effects without adversely impacting normal myocardium, supporting its safety in long-term use.
Ongoing clinical trials are now exploring sildenafil in human DMD and Becker muscular dystrophy patients. If successful, such studies would mark a paradigm shift—transforming a drug associated with erectile enhancement into a cornerstone therapy for inherited muscle diseases. The irony is not lost: a medication developed to treat a condition of excess leisure may ultimately preserve life in one of the most lethal genetic syndromes.
Doxorubicin Cardiotoxicity and the Protective Role of PDE5 Inhibition
Doxorubicin remains a mainstay of cancer chemotherapy but carries the infamous burden of dose-dependent cardiotoxicity. The mechanisms are multifactorial, involving oxidative stress, mitochondrial dysfunction, and apoptosis. Clinical manifestations include left ventricular dysfunction, arrhythmias, and dilated cardiomyopathy. Managing this risk without compromising oncologic efficacy has been an enduring challenge.
Preclinical studies show that sildenafil protects against doxorubicin-induced cardiac injury. By preventing mitochondrial membrane depolarization, inhibiting apoptosis, and preserving contractile proteins, sildenafil maintains left ventricular performance even at cumulative doxorubicin doses known to be cardiotoxic. Tadalafil offers similar protection, up-regulating antioxidant enzymes without interfering with the antitumor activity of doxorubicin. Indeed, co-administration of PDE5 inhibitors with chemotherapy may represent a strategy to balance oncologic potency with cardiac safety.
Intriguingly, sildenafil may also enhance doxorubicin’s anticancer effects. In prostate cancer models, sildenafil sensitized malignant cells to apoptosis while shielding normal cardiomyocytes from oxidative stress. This dual role—oncologic potentiation coupled with cardioprotection—has sparked interest in integrating PDE5 inhibitors into cancer treatment protocols. A once-dismissed “lifestyle drug” may thus become a facilitator of safer, more effective chemotherapy.
cGMP and Oncologic Frontiers
The role of cGMP signaling in oncology extends beyond cardioprotection. PDE5 inhibitors themselves exhibit antiproliferative effects in certain malignancies, inducing caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia and augmenting the permeability of brain tumors to chemotherapeutic agents. By increasing tumor uptake of doxorubicin, sildenafil and vardenafil improved survival in rodent glioma models.
The so-called “Warburg effect,” whereby cancer cells preferentially utilize glycolysis even in oxygen-rich environments, makes them particularly susceptible to oxidative stress. Sildenafil appears to exploit this vulnerability by enhancing reactive oxygen species generation in malignant but not normal cells. In essence, sildenafil tips the redox balance against cancer, compounding the cytotoxicity of conventional agents.
Such findings hint at an unexpected convergence: a cardiovascular drug doubling as an oncologic adjunct. While clinical translation remains in its infancy, the principle is compelling. Manipulating cGMP signaling may simultaneously defend the heart and debilitate the tumor—a duality rare in medicine, and one worth further pursuit.
Conclusions: Sildenafil Beyond Its Origins
From ischemia to hypertrophy, from DMD to doxorubicin cardiotoxicity, cGMP signaling emerges as a unifying thread in diverse cardiac pathologies. PDE5 inhibitors, particularly sildenafil, have moved far beyond their original role as facilitators of penile erection. By sustaining cGMP bioavailability, they unlock a spectrum of protective effects—antiapoptotic, antihypertrophic, antioxidative, and antitumor.
The irony of sildenafil’s journey is almost poetic. What began as an angina drug, reborn as a sexual health revolution, now stands poised to reshape cardiology and oncology. While further clinical trials are essential, the accumulating evidence leaves little doubt: the pharmacological manipulation of cGMP, once an accidental discovery, may become one of the defining therapeutic strategies of modern cardiovascular medicine.
Frequently Asked Questions (FAQ)
1. How does sildenafil protect the heart beyond treating erectile dysfunction?
Sildenafil enhances cGMP signaling by inhibiting PDE5. This activation triggers PKG-dependent pathways that reduce apoptosis, improve mitochondrial function, and prevent maladaptive hypertrophy, all of which contribute to cardioprotection.
2. Can PDE5 inhibitors be safely combined with chemotherapy drugs like doxorubicin?
Yes. Preclinical studies show that sildenafil and tadalafil protect the heart against doxorubicin-induced injury without reducing its anticancer efficacy. In some models, sildenafil even enhances tumor sensitivity to chemotherapy.
3. Is there clinical evidence for sildenafil in treating Duchenne muscular dystrophy?
Animal studies are highly encouraging, and early human trials are underway. Sildenafil restores nitric oxide–cGMP signaling in dystrophin-deficient hearts, reversing established cardiomyopathy within days in preclinical models.
4. Should patients with heart failure routinely receive sildenafil?
Not yet. Clinical trials such as RELAX have produced mixed results. While mechanistic rationale is strong, widespread use awaits more definitive evidence from large, controlled human studies.